We have prepared and isolated in solid form two types of polymers formed between iron (II) complexes ([FePc(COOH)8] and [FePc(CN)8]) and two bidentate ligands [trans-1,2-bis (4-pyridyl) ethylene (bpe); trans-1,2-bis (4-pyridyl) ethane (bpa)]. The electronic and vibrational absorption spectra of these complexes are discussed in comparison with those of previous work on [FePcL2]n polymers with the same ligands. Infrared spectrometry shows a modulation in the intensities of certain characteristic bands of the complexes, reflecting a reorganization of the structure of these compounds through the formation of polymers and, above all, the emergence of new vibration bands attributable to the ligands. In electron absorption spectrometry, our results confirm those already available in the literature with the [FePcL2]n series. The presence of the bpa ligand causes each macrocycle of the polymer to behave independently. In contrast, the bpe ligand induces a perfect linear connection between the macrocycles due to its alkene function, which allows electrons to move easily along the polymer chain. The presence of peripheral groups (COOH and CN) provides a novel result because they strongly influence not only the energy of the π→π* band, but especially that of the central metal-axial ligand charge transfer band (CT Fe→L). These charge transfers are responsible for the conductive properties of these compounds.
| Published in | Science Journal of Chemistry (Volume 14, Issue 1) |
| DOI | 10.11648/j.sjc.20261401.13 |
| Page(s) | 25-37 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Iron Phthalocyanine, Bpe, Bpa, UV-Visible, IR
Compounds |
|
|
|
|---|---|---|---|
[FePc] | 649 | - | - |
[FePc(COOH)8] | 682 | - | - |
[FePc(CN)8] | 686 | - | - |
Compounds |
|
|
|
|---|---|---|---|
[FePc] | 649 | - | - |
[FePc(Py)2] | 653 | - | 406 |
[FePc(4CNPy)2] | 649 | 497 | 397 |
[FePc(bpa)2]n | 652 | - | 405 |
[FePc(bpe)2]n | 649 | 575 | 400 |
Compounds |
|
|
|
|---|---|---|---|
[FePc(COOH)8] | 682 | - | - |
[FePc(COOH)8(Py)2] | 673 | - | 424 |
[FePc(COOH)8(4CNPy)2] | 673 | 540e | 418 |
[FePc(COOH)8(bpa)2]n | 672 | - | 425 |
[FePc(COOH)8(bpe)2]n | 671 | 552 | 425 |
Compounds |
|
|
|
|---|---|---|---|
[FePc(CN)8] | 684 | - | - |
[FePc(CN)8(Py)2] | 678 | - | 417 |
[FePc(CN)8(4CNPy)2] | 697 | 557 | 426 |
[FePc(CN)8(bpa)2]n | 683 | - | 415 |
[FePc(CN)8(bpe)2]n | 694 | 567 | 423 e |
[FePc(COOH)8] | [FePc(COOH)8Py2] | [FePc(COOH)8(bpa)2]n | [FePc(COOH)8(4CNPy)2] | [FePc(COOH)8(bpe)2]n | Attributions |
|---|---|---|---|---|---|
430 f | 431 f | 440 f | ΦC-C deformation du macrocycle | ||
482 m | 458 m | 466 f | 433 m | 452 f | |
501 m | 501 m | 506 f | 501 f | 496 m | |
543 m | 547 m | 555 m | 538 m | 546 m | |
581 m | 632 F | 590 m | 600 f | 599 m | |
627 m | 630 F | 632 F | 627 F | ||
662 m | 667 F | ||||
676 m | 688 F-697 F | 696 m | 695 m | ||
732 TF | 734 F | 732 F | 733 TF | 731TF | C-H |
749 F | 750 f | 799 m | 750 F | 749 m | C–H |
799 F | 801 f | 800 m | 799 m | ||
825 m | 823 m | 826 m | 824 m | ||
918 F | 921 F | 916 F | 923 F | 916 m | M-N |
1000 F | 1008 F | 1006 m | 1000 F | 1005 m | |
1090 TF | 1090 F | 1088 TF | 1088 TF | 1086 F | βC-H |
1143 f | 1150 m | 1153 m | 1151 m | ||
1163 f | - | 1155 m | |||
1180 f | - | ||||
1233 F | 1237 m | 1236 m | 1242 m | 1237 m | C-O |
1268 F | 1272 F | 1268 F | 1270 F | 1264 TF | C-C |
1317 m | 1319 m | 1318 f | 1316 m | 1317 f | C-N |
1369 m | 1374 m | 1376 F | 1369 m | 1368 m | |
1437 m | 1452 F | 1450 F | 1450 m | M-N | |
1450 m | 1498 m | 1514 F | 1450 F | 1515 F | Vibration C=N du cycle |
1516 m | 1516 m | 1582 F | 1516 m | 1582 F | |
1586 F | 1583 m | 1586 m | |||
1628 m | 1635 f | 1637 F | |||
1703 TF | 1705 TF | 1703 TF | 1704 TF | 1697 TF | C=O |
2500-3500 l | 2500-3500 l | 2500-3500 l | 2500-3500 l | 2500-3500 l | νO-H |
[FePc(CN)8] | [FePc(CN)8Py2] | [FePc(CN)8(bpa)2]n]n | [FePc(CN)8(4CNPy)2] | [FePc(CN)8(bpe)2]n | Attributions |
|---|---|---|---|---|---|
480 f | - | 477 f | - | 533 F | ΦC-C deformatio n du macrocycle |
529 TF | - | 529 TF | - | 613 m | |
- | - | - | - | - | |
636 f | - | 637 f | - | ||
684 f | - | 684 f | 672 f | ||
704 f | - | 702 f | - | 705 m | |
721 F | 724 TF | 721 F | 734 TF | 721 m | νC–H |
759 F | - | 757 F | - | 761 F | C–H |
800 F | 808 m | 800 F | 818 F | 800 F | C–H |
872 m | - | 872 m | - | - | |
915 m | 923 m | 920 F | 933 F | 909 f | M-N |
1028 F | 1038 m | 1028 F | 1047 f | 1051 m | |
1096 TF | 1112 TF | 1096 TF | 1121 TF | 1106 TF | βC-H |
1164 m | 1173 f | 1164 m | 1182 f | 1142 m | |
1268 F | 1276 TF | 1269 F | 1276 TF | 1267 m | C-C |
1310 TF | 1309 TF | 1310 TF | 1318 TF | 1318 F | C-N |
1412 m | - | 1412 m | - | 1415 m | M-N Cycle vibration C=N |
1443 m | 1452 m | 1445 m | 1465 F | 1443 f | |
1519 F | 1518 f | 1519 F | 1529 m | 1515 F | |
- | 1549 f | - | 1560 f | 1569 f | |
1573 m | - | 1571 m | - | - | |
- | 1620 f | - | 1628 f | - | |
- | 1659 f | - | 1667 f | - | |
1715 m | 1725 m | 1710 m | 1736 m | 1724 f | C=N bonded |
2223 F | 2226 F | 2223 F | 2236 TF | 2226 F | νC≡N |
- | - | 2900-2987 m | 2900-2990 m | νCH2; νCH |
Bpe | Trans-1,2-bis (4-pyridyl) Ethylene |
Bpa | Trans-1,2-bis (4-pyridyl) Ethane |
IR | Infrared |
DMSO | Dimethyl Sulfoxide |
Py | Pyridine |
4CNPy | 4-CNpyridine |
ATR | Attenuated Total Reflectance |
CT | Charge Transfert |
| [1] | Karifa, B., Guy, V. O., Georges, T., David, B. UV-VISIBLE AND IR spectra of iron(II) phthalocyanine polymer complexes linked by bis-pyridinato ligands. Polyhedron. 1990, 9(8), 1087-1090. |
| [2] |
Soo, J. K., Michiko, M., Kiyotaka, S. Synthesis and electrical properties of one-dimensional octacyanometallophtalocyanine (M= Fe, Co) polymers. Journal of the Porphyrins and Phthalocyanines. 2000, 4(1), 136-144.
https://doi.org/10.1002/(SICI)1099-1409(200001/02)4:13.0.CO;2-J |
| [3] | Leonardo, A., Chiara, T., Brian, S., Alberto, C. Molecular devices based on the mechanical bond: recent advances. Coordination Chemistry Reviews. 2006, 553, 217583. |
| [4] | Michael, H., Uwe, K., Hans-joachim, S. Iodine-doped bridged phthalocyaninatoiron (II) And-ruthenium(II) Compounds. Synthetic Metals. 1987, 20(3), 347-356. |
| [5] | Sami, A., Néji, B., Bassem,: Phthalocyanines: Syntheses and Proprietes. Moroccan Journal of Heterocyclic Chemistry. 2021, 20(1), 1-14. |
| [6] | Mendizabal, F., Olea-Azar, C., Zapata-Torres, G., Eisner, F. Metallomacrocycle complex bridged polymers: electronic structures of–[MacM (L)]n. Journal of Molecular Structure THEOCHEM. 2001, 543(1-3), 23-37. |
| [7] | Schick, G. A., Bocian, D. F. Resonance Raman studies of bis (pyridine) adducts of iron (II), ruthenium (II), and osmium (II) octaethylporphyrins. Effects of heavy-metal substitution on porphyrin and axial-ligand vibrational and electronic properties. Journal of the American Chemical Society. 1984, 106(6), 1682-1694. |
| [8] | Schneider, O., Hanack, M. Phthalocyanine iron with pyrazine as a bidentate bridging ligand. Angewandte Chemie. 1980, 92(5), 391-393. |
| [9] | Danilo D, Michael H. Phthalocyanines as materials for advanced technologies: some examples. Journal of the Porphyrins and Phthalocyanines. 2004, 8(7), 915-933. |
| [10] | Nakagawa, M., Rikukawa, M., Sanui, K., Ogata, N. Synthesis, electrochemical, and electrical properties of (phthalocyaninato) iron complexes with azopyridines. Synthetic metals. 1997, 84(1-3), 391-392. |
| [11] | Victor, N. N., Evgeny, A. L. Synthesis of substituted phthalocyanines. ARKIVOC. 2010, (2010) 1, 136-208. |
| [12] | Nitiema, W. K. G. A., Lassane, T., Bertrand, O., Bayo-Bangoura, M., Holade, Y., Bayo, K. Preparation, vibrational and electronic studies of octasubstituted gold phthalocyanine complexes and their derivatives bearing axial pyridine and substituted pyridine ligands. Journal of the West African Society of ChemistryPreparation, vibrational and electronic studies of octasubstituted gold phthalocyanine complexes and their derivatives bearing axial pyridine and substituted pyridine ligands. Journal of the West African Society of Chemistry. 2024, 53, 74-84. |
| [13] |
Tarpaga, L., Bayo-Bangoura, M., Ouédraogo, S., Bayo, K. Preparation, vibrational and electronic studies of hexacoordinated complexes of iron octocarboxyphthalocyanine with pyridine axial ligands and substituted pyridines, phosphine and phosphites. Asian Journal of Science and Technology. 2018, 9(5), 8185-8192.
https://www.journalajst.com/sites/default/files/issues-pdf/6052.pdf |
| [14] | Zanguina, A., Bayo, K., Bayo-Bangoura, M., Ouédraogo, G. V. IR and UV-visible spectra of iron (II) phthalocyanine complexes with phosphine or phosphite. Bulletin of the Chemical Society of Ethiopia. 2002, 16(1), 73-79. |
| [15] | Sakamoto, K., Ohno-okumura, E. Syntheses and functional properties of phthalocyanines. Materials. 2009, 2(3), 1127-1179. |
| [16] | Xuezheng, Y., Shoujuan, L., Shishan, X., Shuai, C., Xiaoli, Z., Xilin, S., Tianrong, Z., Xiaoliang, Z., Dongjiang, Y. Coupling of iron phthalocyanine at carbon defect site via π-π stacking for enhanced oxygen reduction reaction. Applied Catalysis B: Environment and Energy. 2021, 280, 119437. |
| [17] | Jinping, W., Xiaoming, Z., Yi, Z., Zhen, P., Chunzhen, Z., Fenglian, Z. Structural design of porous organic polymers to mitigate π-stacking-induced quenching in porphyrin/phthalocyanine photosensitizers for enhanced antibacterial activity. RSC Advances. 2025, 15(7), 48604-48627. |
| [18] | Ouedraogo, G. V., More, C., Richard, Y., Benlian, D. Charge-transfer and Moessbauer spectra of axially substituted iron (II) phthalocyanines. Inorganic Chemistry. 1981, 20(12), 4387-4393. |
| [19] | Dustin, E. N, Laura, S. F., Briana, R. S., Victor, N. N. Charge-Transfer Spectroscopy of Bisaxially Coordinated Iron(II) Phthalocyanines through the Prism of the Lever’s EL Parameters Scale, MCD Spectroscopy, and TDDFT Calculations. Inorganic Chemistry. 2022, 61(21), 8250-8266. |
| [20] | Fierro, C., Anderson, A. B., Scherson, D. A. Electron donor-acceptor properties of porphyrins, phtalocyanines, and related ring chelates: a molecular orbital study. Journal of Physical and. Chemistry. 1988, 92(24), 6902-6907. |
| [21] | Fickling, M. M., Fischer, A., Mann, B. R., Packer, J., Vaughan, J. Hammett substituent constants for electron-withdrawing substituents: Dissociation of phenols, anilinium ions and dimethylanilinium ions. Journal of the American Chemical Society. 1959, 81(16), 4226-4230. |
| [22] | Kadish, K. M., Smith, K. M., Guilard, R. Structure-Property Relationships in Porphyrin Systems. The Porphyrin Handbook: Multporphyrins, Multiphthalocyanines and Arrays. 2012. 19(1), 1-354. |
| [23] | Benjamin, W. G., Mehul, P., Xiaowen, X. A Threshold for Charge Transfer in Aromatic Interactions? A Quantitative Study of π-Stacking Interactions. Journal of Organic Chemistry. 2005, 70(25), 10532-10537. |
| [24] | Inabe, T., Tajima, H. Phthalocyanines versatile components of molecular conductors. Chemical Reviews. 2004, 104(11), 5503-5534. |
| [25] | Sanda-Bawa, A., Bayo-Bangoura, M., Ouemega, B., Bayo, K. Preparation and vibrational and electronic studies of gold phthalocyanine complexes with axial pyridine and substituted pyridine ligands. International Journal of biological and chemical sciences. 2018, 12(3), 1516-1527. |
| [26] | Emmanuël, L. Spectrometric identification of organic compounds: translation of the 5th American edition. Paris: Deboeck Université 2004, 142 p. |
| [27] | Richard, N. A. 2001. Interpreting infrared, raman, and nuclear magnetic resonance spectra. Elsevier; 2001, 1. |
| [28] | Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd edition. Wiley; 2004, 368 p. |
| [29] | Barbara, H. S. Infrared spectroscopy: fundamentals and applications. John Wiley & Sons, Ltd; 2004; 231 p. |
APA Style
Tarpaga, L., Nitiema, W. G. A., Ouedraogo, S., Ouemega, B., Sessouma, B., et al. (2026). Synthesis and Spectrometric Study of New Iron Phthalocyanine Polymers: Influence of Peripheral COOH and CN Groups on Vibrational and Electronic Properties. Science Journal of Chemistry, 14(1), 25-37. https://doi.org/10.11648/j.sjc.20261401.13
ACS Style
Tarpaga, L.; Nitiema, W. G. A.; Ouedraogo, S.; Ouemega, B.; Sessouma, B., et al. Synthesis and Spectrometric Study of New Iron Phthalocyanine Polymers: Influence of Peripheral COOH and CN Groups on Vibrational and Electronic Properties. Sci. J. Chem. 2026, 14(1), 25-37. doi: 10.11648/j.sjc.20261401.13
AMA Style
Tarpaga L, Nitiema WGA, Ouedraogo S, Ouemega B, Sessouma B, et al. Synthesis and Spectrometric Study of New Iron Phthalocyanine Polymers: Influence of Peripheral COOH and CN Groups on Vibrational and Electronic Properties. Sci J Chem. 2026;14(1):25-37. doi: 10.11648/j.sjc.20261401.13
@article{10.11648/j.sjc.20261401.13,
author = {Lassane Tarpaga and Wend-Kuny Guy Aristide Nitiema and Seydou Ouedraogo and Bertrand Ouemega and Bintou Sessouma and Mabinty Bayo-Bangoura and Karifa Bayo},
title = {Synthesis and Spectrometric Study of New Iron Phthalocyanine Polymers: Influence of Peripheral COOH and CN Groups on Vibrational and Electronic Properties},
journal = {Science Journal of Chemistry},
volume = {14},
number = {1},
pages = {25-37},
doi = {10.11648/j.sjc.20261401.13},
url = {https://doi.org/10.11648/j.sjc.20261401.13},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.sjc.20261401.13},
abstract = {We have prepared and isolated in solid form two types of polymers formed between iron (II) complexes ([FePc(COOH)8] and [FePc(CN)8]) and two bidentate ligands [trans-1,2-bis (4-pyridyl) ethylene (bpe); trans-1,2-bis (4-pyridyl) ethane (bpa)]. The electronic and vibrational absorption spectra of these complexes are discussed in comparison with those of previous work on [FePcL2]n polymers with the same ligands. Infrared spectrometry shows a modulation in the intensities of certain characteristic bands of the complexes, reflecting a reorganization of the structure of these compounds through the formation of polymers and, above all, the emergence of new vibration bands attributable to the ligands. In electron absorption spectrometry, our results confirm those already available in the literature with the [FePcL2]n series. The presence of the bpa ligand causes each macrocycle of the polymer to behave independently. In contrast, the bpe ligand induces a perfect linear connection between the macrocycles due to its alkene function, which allows electrons to move easily along the polymer chain. The presence of peripheral groups (COOH and CN) provides a novel result because they strongly influence not only the energy of the π→π* band, but especially that of the central metal-axial ligand charge transfer band (CT Fe→L). These charge transfers are responsible for the conductive properties of these compounds.},
year = {2026}
}
TY - JOUR T1 - Synthesis and Spectrometric Study of New Iron Phthalocyanine Polymers: Influence of Peripheral COOH and CN Groups on Vibrational and Electronic Properties AU - Lassane Tarpaga AU - Wend-Kuny Guy Aristide Nitiema AU - Seydou Ouedraogo AU - Bertrand Ouemega AU - Bintou Sessouma AU - Mabinty Bayo-Bangoura AU - Karifa Bayo Y1 - 2026/02/27 PY - 2026 N1 - https://doi.org/10.11648/j.sjc.20261401.13 DO - 10.11648/j.sjc.20261401.13 T2 - Science Journal of Chemistry JF - Science Journal of Chemistry JO - Science Journal of Chemistry SP - 25 EP - 37 PB - Science Publishing Group SN - 2330-099X UR - https://doi.org/10.11648/j.sjc.20261401.13 AB - We have prepared and isolated in solid form two types of polymers formed between iron (II) complexes ([FePc(COOH)8] and [FePc(CN)8]) and two bidentate ligands [trans-1,2-bis (4-pyridyl) ethylene (bpe); trans-1,2-bis (4-pyridyl) ethane (bpa)]. The electronic and vibrational absorption spectra of these complexes are discussed in comparison with those of previous work on [FePcL2]n polymers with the same ligands. Infrared spectrometry shows a modulation in the intensities of certain characteristic bands of the complexes, reflecting a reorganization of the structure of these compounds through the formation of polymers and, above all, the emergence of new vibration bands attributable to the ligands. In electron absorption spectrometry, our results confirm those already available in the literature with the [FePcL2]n series. The presence of the bpa ligand causes each macrocycle of the polymer to behave independently. In contrast, the bpe ligand induces a perfect linear connection between the macrocycles due to its alkene function, which allows electrons to move easily along the polymer chain. The presence of peripheral groups (COOH and CN) provides a novel result because they strongly influence not only the energy of the π→π* band, but especially that of the central metal-axial ligand charge transfer band (CT Fe→L). These charge transfers are responsible for the conductive properties of these compounds. VL - 14 IS - 1 ER -
Laboratory of Molecular Chemistry and Materials, Joseph KI-ZERBO University, Ouagadougou, Burkina Faso
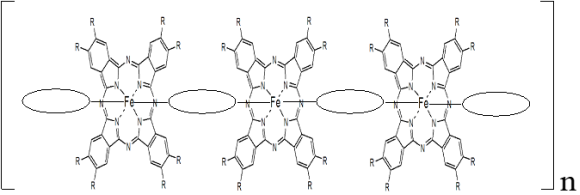
Figure 1. Representation of polymer iron phthalocyanine with R = COOH or CN and![]() .
.
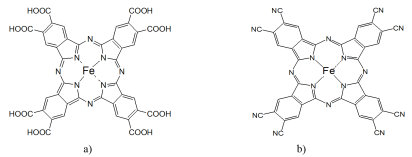
Figure 2. (a) Iron octacarboxylate phthalocyanine [FePc(COOH)8] and (b) iron octacyanine phthalocyanine [FePc(CN)8].
Figure 3. Structure of pyridine.
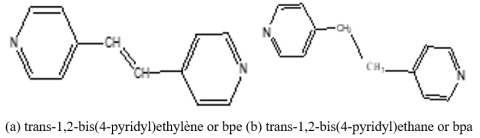
Figure 4. Structure of (a) bpe and (b) bpa.
Figure 5. Structure of 4-CNpyridine.
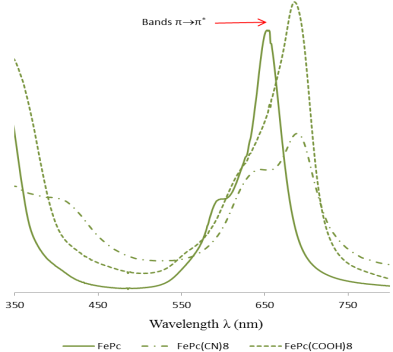
Figure 6. UV-visible spectra of FePc, [FePc(COOH)8] and [FePc(CN)8] in DMSO solution.
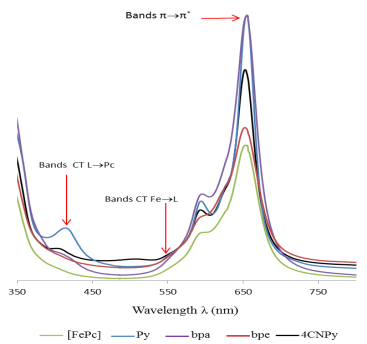
Figure 7. UV-visible spectra of FePc and the reaction compounds of [FePc] with pyridine and its derivatives (bpa, bpe, 4CNPy) in solution in DMSO.
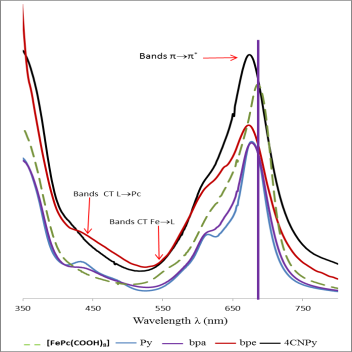
Figure 8. UV-visible spectra of [FePc(COOH)8] and the reaction compounds of [FePc(COOH)8] with pyridine and its derivatives (bpa, bpe, 4CNPy) in DMSO solution.
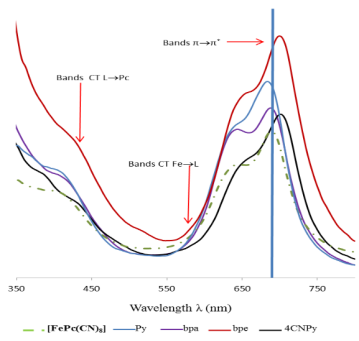
Figure 9. UV-visible spectra of [FePc(CN)8] and the reaction compounds of [FePc(CN)8] with pyridine and its derivatives (bpa, bpe, 4CNPy) in solution in DMSO.
Information

